
- |<
- <
- 1
- >
- >|
-
Kentaro Kawai2024 年 72 巻 9 号 p. 775
発行日: 2024/09/01
公開日: 2024/09/01
ジャーナル オープンアクセス HTMLPDF形式でダウンロード (232K) HTML形式で全画面表示
-
Shinya Nakamura, Keiji Nishiwaki, Masato Tsuyuguchi, Takayoshi Kinoshi ...2024 年 72 巻 9 号 p. 776-780
発行日: 2024/09/01
公開日: 2024/09/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLProtein kinase CK2 type α (CK2α) inhibitors are expected to be a new anticancer drug and a treatment for nephritis. Virtual screening for CK2α inhibitors has been conducted and active compounds with various scaffolds have been obtained. Research on compound optimization is currently in progress for some of them with the aim of improving their activity. This process involves the combination of various computational chemistry methods and crystal analyses. In this review, case studies of structure-based compound designs that have efficiently improved the activity of screening hit compounds, including compounds with a thiadiazole ring and a purine scaffold, are introduced.
抄録全体を表示PDF形式でダウンロード (1284K) HTML形式で全画面表示 -
Daisuke Takaya2024 年 72 巻 9 号 p. 781-786
発行日: 2024/09/01
公開日: 2024/09/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLOwing to the increasing use of computers, computer-aided drug design (CADD) has become an essential component of drug discovery research. In structure-based drug design (SBDD), including inhibitor design and in silico screening of drug target molecules, concordance with wet experimental data is important to provide insights on unique perspectives derived from calculations. Fragment molecular orbital (FMO) method is a quantum chemical method that facilitates precise energy calculations. Fragmentation method makes it possible to apply the quantum chemical method to biological macromolecules for energy calculation based on the electron behavior. Furthermore, interaction energies calculated on a residue-by-residue basis via fragmentation aid in the analysis of interactions between the target and ligand molecule residues and molecular design. In this review, we outline the recent developments in SBDD and FMO methods and highlight the prospects of developing machine learning approaches for large computational data using the FMO method.
抄録全体を表示PDF形式でダウンロード (3734K) HTML形式で全画面表示 -
Norihito Kawashita2024 年 72 巻 9 号 p. 787-793
発行日: 2024/09/01
公開日: 2024/09/01
 ジャーナル オープンアクセス HTML
ジャーナル オープンアクセス HTMLThe use of computational methods in drug discovery research has increased substantially in recent years. Computational chemistry techniques, such as quantum chemical calculations and molecular dynamics simulations, continue to be widely used. In this review, we focused on drug discovery-related studies that employ fragment molecular orbital methods. Furthermore, we focused on inhibitor discovery, protein–protein interaction analysis, including antigen–antibody interaction analysis, and integration with molecular dynamics simulations.
抄録全体を表示PDF形式でダウンロード (464K) HTML形式で全画面表示 -
 Kentaro Kawai, Yukiko Karuo, Atsushi Tarui, Kazuyuki Sato, Makoto Kata ...2024 年 72 巻 9 号 p. 794-799
Kentaro Kawai, Yukiko Karuo, Atsushi Tarui, Kazuyuki Sato, Makoto Kata ...2024 年 72 巻 9 号 p. 794-799
発行日: 2024/09/01
公開日: 2024/09/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLRecently, remarkable progress has been achieved in artificial intelligence (AI), including machine learning. Various AI models have been proposed for drug discovery, including the design of small molecules, activity prediction, and three-dimensional (3D) structure prediction of proteins. AI consists of diverse elements, including information retrieval and machine learning, and can be used in a wide range of drug discovery scenarios. In this review, we focused on AI for small-molecule drug discovery with respect to molecular design, activity prediction, and prediction of the binding poses of compounds to target molecules. We also discussed the applications of AI in academic drug discovery.
抄録全体を表示Editor's pickProductivity in drug discovery is expected to increase through greater use of in silico technology. The authors have developed AI models applicable to a variety of drug discovery tasks, including molecular design, activity prediction and side-effect profiling. The characteristics of training data in the context of drug discovery are also discussed, providing insights for future AI development. The authors also perform wet experiments in addition to dry research. They present examples of using in silico technology for drug discovery, including developing pharmacokinetic enhancers and drug design against multidrug-resistant bacteria.
PDF形式でダウンロード (1586K) HTML形式で全画面表示
-
Akira Kotani, Koichi Machida, Ryo Watanabe, Hideki Hakamata2024 年 72 巻 9 号 p. 800-803
発行日: 2024/09/03
公開日: 2024/09/03
 ジャーナル オープンアクセス HTML
ジャーナル オープンアクセス HTML
電子付録A noise filter, which is usually attached to a detector for chromatography, was applied for the improvement of a signal-to-noise ratio (S/N) on a chromatogram. The objective of this paper is to elucidate the effect of noise filtering in an UV detector of ultra HPLC (UHPLC) on the statistical reliability of chemometrically evaluated repeatability by the function of mutual information (FUMI) theory. To examine the statistical reliability of chemometrically evaluated repeatability in the UHPLC system associated with noise filtering, the standard deviation (SD) values of the area in baseline fluctuations with peak region k (s(k)) were obtained from six chromatograms with noise filtering. Further, the average of s(k) values (σ̂) was calculated from the s(k) values (n = 6) to be alternatively applied as the population SD. All s(k)/σ̂ values were within the 95% confidence intervals (CIs) at the freedom degree of 50, indicating the chemometrically estimated relative SD (RSD) of a peak area and RSD by repeated measurements of at least 50 times had equivalent reliability.
抄録全体を表示Editor's pickRepeatability is a significant parameter, expressed as the relative standard deviation (RSD) of measured values, applied to validate the performance of a UHPLC system. The authors proposed a chemometric tool to estimate the RSD of the peak area obtained from the UHPLC equipped with a noise filter, and the RSDs estimated by this tool were demonstrated to be more reliable than those by 50 repetitive measurements. Using the chemometric tool, the resources needed to evaluate repeatability can be reduced, and thus the efficiency of repeatability evaluation will be remarkably improved in a UHPLC analysis.
PDF形式でダウンロード (1046K) HTML形式で全画面表示
-
Akihiro Ambo, Shiho Ohno, Yoshiki Yamaguchi, Masayuki Seki2024 年 72 巻 9 号 p. 804-809
発行日: 2024/09/10
公開日: 2024/09/10
 ジャーナル オープンアクセス HTML
ジャーナル オープンアクセス HTML
電子付録Protein-based enzymes are among the most efficient catalysts on our planet. A common feature of protein enzymes is that all catalytic amino acids occupy a limited, narrow space and face each other. In this study, we created a theoretical novel biomimetic molecule containing different multiple catalytic peptides. Although single peptides are far less catalytically efficient than protein enzymes, Octopus-arms-mimicking biomolecules containing eight different peptides (Octopuzymes) can efficiently catalyze organic reactions. Since structural information for extant protein enzymes, predicted enzymes based on genome data, and artificially designed enzymes is available for designing Octopuzymes, they could in theory mimic all protein enzyme reactions on our planet. Moreover, besides L-amino acids, peptides can contain D-amino acids, non-natural amino acids, chemically modified amino acids, nucleotides, vitamins, and manmade catalysts, leading to a huge expansion of catalytic space compared with extant protein enzymes. Once a reaction catalyzed by an Octopuzyme is defined, it could be rapidly evolvable via multiple amino acid substitutions on the eight peptides of Octopuzymes.
抄録全体を表示PDF形式でダウンロード (2707K) HTML形式で全画面表示 -
Mizuki Sugimoto, Eita Sasaki, Hisashi Ohno, Takayuki Ikeno, Sota Yamad ...2024 年 72 巻 9 号 p. 810-816
発行日: 2024/09/20
公開日: 2024/09/20
 ジャーナル オープンアクセス HTML
ジャーナル オープンアクセス HTML
電子付録Twisted intramolecular charge transfer (TICT) is a phenomenon involving intramolecular charge transfer together with intramolecular rotation upon photoexcitation, and in general this excited state of fluorescent dyes undergoes non-radiative decay (producing no fluorescence). We recently discovered that the magnitude of TICT in rhodamine derivatives could be regulated by altering the size of the substituents on the xanthene moiety, generating differing degrees of intramolecular steric repulsion. To further illustrate the usefulness and generality of this strategy, we describe here an application of quinone methide chemistry, which is widely used as a fluorescence off/on switching reaction for fluorescence probes detecting enzymatic activity, to construct a steric repulsion-induced (sr)-TICT-based fluorescence probe targeting nitroreductase (NTR) activity. The developed probe was almost non-fluorescent in phosphate-buffered saline (PBS) due to strong induction of the TICT state. On the other hand, when the probe was incubated with NTR and nicotinamide adenine dinucleotide (NADH), a large fluorescence increase was observed over time. We confirmed that the enzymatic reaction proceeded as expected, i.e., the nitro group of the probe was reduced to the corresponding amino group, followed by spontaneous elimination of iminoquinone methide. These results suggest that our simple design strategy based on the sr-TICT mechanism, i.e., controlling intramolecular steric repulsion, would be applicable to the development of fluorescence probes for a variety of enzymes.
抄録全体を表示Editor's pickThis report described the usefulness of the design strategy for fluorescence probes that controls intramolecular steric repulsion, previously reported by the authors, i.e., the steric repulsion-induced twisted intramolecular charge transfer (sr-TICT) mechanism. To demonstrate the utility of this strategy, the authors describe an application of quinone methide chemistry, which is widely used as a fluorescence off/on switching reaction in fluorescence probes for enzymatic activity detection, to construct a fluorescence probe targeting nitroreductase (NTR) activity. This research suggests that the sr-TICT mechanism would be useful for the development of fluorescence probes for a variety of enzymes.
PDF形式でダウンロード (1170K) HTML形式で全画面表示 -
Kenta Fujinuma, Shota Okada, Kyu Hayashi, Masataka Ito, Hironori Suzuk ...2024 年 72 巻 9 号 p. 817-825
発行日: 2024/09/20
公開日: 2024/09/20
 ジャーナル オープンアクセス HTML
ジャーナル オープンアクセス HTML
電子付録The triboelectric properties of active pharmaceutical ingredients (APIs) contribute to problems during the manufacturing of pharmaceuticals. However, the triboelectric properties of APIs have not been comprehensively characterized. In this study, the effect of salt formulation on the triboelectric properties of APIs was investigated. The triboelectric properties of three groups of amines, namely tertiary amines, purine bases, and amino acids, and their hydrochlorides were evaluated using a suction-type Faraday cage meter. Most of the hydrochloride salts exhibited more negative charges than the corresponding free bases, and the degree by which the triboelectric property changed upon hydrochlorination depended on the structural groups of the compounds. In the case of tertiary amines, the change in the zero-charge margin upon hydrochlorination was negatively correlated with the zero-charge margin of the free base. In contrast, hydrochlorination of the amino acids led to a significant change in the zero-charge margin. In most cases, salt formation also affected the triboelectric properties of API powders. Controlling the triboelectric properties of APIs solves various problems caused by the electrification of raw material powders and granules during the production of pharmaceuticals, thereby increasing the quality of produced pharmaceuticals.
抄録全体を表示PDF形式でダウンロード (1587K) HTML形式で全画面表示 -
Kenichi Matsuda, Shinya Niikura, Rintaro Ichihara, Kei Fujita, Anna M. ...2024 年 72 巻 9 号 p. 826-830
発行日: 2024/09/20
公開日: 2024/09/20
 ジャーナル オープンアクセス HTML
ジャーナル オープンアクセス HTML
電子付録Surugamides are a group of non-ribosomal peptides produced by Streptomyces spp. Several derivatives possess acyl groups, which are proposed to be attached to a lysine side chain after backbone-macrocyclization during biosynthesis. To date, five different acyl groups have been identified in nature, yet their impacts on biological activity remain underexplored. Here we synthesized surugamide B derivatives with varied acyl moieties. Biological evaluations revealed that larger hydrophobic acyl groups on lysine ε-NH2 enhance cytotoxicity.
抄録全体を表示Editor's pickSurugamides are cyclic octapeptides originally isolated from Streptomyces as cathepsin B inhibitors. Subsequent studies have shown that derivatives featuring an acyl group on the e-amino group of the lysine residue exhibit higher activity compared to non-acylated surugamides in cell-based or whole-organism assays. This study evaluated the cytotoxicity profiles of four synthetic acyl surugamides, revealing that larger hydrophobic acyl groups on the lysine e-amino group enhance cytotoxicity. It also identified new derivatives with sub-micromolar potencies and demonstrated that the appropriate size of the acyl substituent is crucial for activity.
PDF形式でダウンロード (756K) HTML形式で全画面表示 -
Kyohei Muguruma, Akane Fukuda, Hayate Shida, Akihiro Taguchi, Kentaro ...2024 年 72 巻 9 号 p. 831-837
発行日: 2024/09/20
公開日: 2024/09/20
 ジャーナル オープンアクセス HTML
ジャーナル オープンアクセス HTML
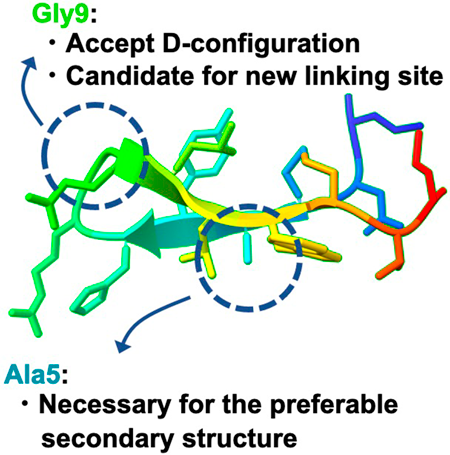
電子付録Mid-sized cyclic peptides are a promising modality for modern drug discovery. Their larger interaction area coupled with an appropriate secondary structure is more suitable than small molecules for binding to the target protein. In this study, we conducted a structure derivatization of an immunoglobulin G (IgG)-binding peptide (15-IgBP), a β-hairpin-like cyclic peptide with a twisted β-strand and assessed the effect of the secondary structure on IgG-binding activity using circular dichroism (CD) spectra analysis. As a result, derivatization at the Ala5 and Gly9 positions affected the secondary structure of 15-IgBP, in particular the appearance of a small positive peak in the 220–240 nm region characteristic of 15-IgBP in the CD spectrum. Maintaining this peak at a moderate level may be important for the expression of IgG binding activity. We found the small methyl group at Ala5 to be crucial for retaining the preferred secondary structure; we also found Gly9 could be replaced by D-amino acids. By integrating these findings with previous results of the structure–activity relationship, we obtained four potent affinity peptides for IgG binding (Kd = 4.24–5.85 nM). Furthermore, we found the Gly9 position can be substituted for D-Lys. This is a new potential site for attaching functional units for conjugation with IgG for the preparation of homogeneous antibody–drug conjugates.
抄録全体を表示Editor's pick[Highlighted Paper selected by Editor-in-Chief]
Affinity peptides that target the fragment crystallizable (Fc) region of IgG (IgG-binding peptides) are widely employed in pharmaceutical applications. The authors conducted structural derivatization of an IgG-binding peptide – specifically, a hairpin-like cyclic peptide featuring a twisted beta-strand – and evaluated the effect of its secondary structure on IgG-binding activity using circular dichroism (CD) spectroscopy. Through the integration of both current and previous findings, four potent IgG-binding affinity peptides were identified. This research has significant potential for the application of IgG-binding peptides, particularly in the development of homogeneously modified antibody-drug conjugates (ADCs).PDF形式でダウンロード (2671K) HTML形式で全画面表示 -
Kenta Ando, Hiromasa Uchiyama, Katsuhiko Minoura, Kazunori Kadota, Yui ...2024 年 72 巻 9 号 p. 838-844
発行日: 2024/09/27
公開日: 2024/09/27
 ジャーナル オープンアクセス HTML
ジャーナル オープンアクセス HTML
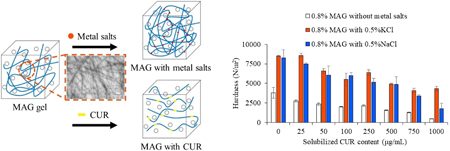
電子付録Monoammonium glycyrrhizic acid (MAG), a glycyrrhizic acid monoammonium salt, is a naturally derived low-molecular-weight gelling agent with surface-active properties. It has the capacity to individually facilitate the preparation of gel-solubilized drugs. As MAG is an anionic surfactant with carboxyl groups, the addition of counterions may affect micelle formation and gelation. In this study, the solubilization and gelling properties of MAG were investigated following the addition of metal salts (NaCl and KCl). The addition of metal salts resulted in a decrease in the critical micelle concentration and an increase in gel hardness. Supersaturation of curcumin (CUR) was maintained by the addition of metal salts because of increased micelle number and viscosity. When the gel hardness was compared between formulations with and without CUR, a significant reduction in hardness was observed with the solubilization of CUR. The addition of KCl prevented the decrease in the hardness of gels containing CUR compared to the addition of NaCl. Put together, the addition of metal salts had a noteworthy impact on micelle and gel formation of MAG. In particular, the addition of KCl was more effective in the preparation of gel-solubilized CUR.
抄録全体を表示PDF形式でダウンロード (4920K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|