Abstract
The goal of drug design is to discover molecular structures that have suitable pharmacological properties in vast chemical space. In recent years, the use of deep generative models (DGMs) is getting a lot of attention as an effective method of generating new molecules with desired properties. However, most of the properties do not have three-dimensional (3D) information, such as shape and pharmacophore. In drug discovery, pharmacophores are valuable clues in finding active compounds. In this study, we propose a computational strategy based on deep reinforcement learning for generating molecular structures with a desired pharmacophore. In addition, to extract selective molecules against a target protein, chemical genomics-based virtual screening (CGBVS) is used as post-processing method of deep reinforcement learning. As an example study, we have employed this strategy to generate molecular structures of selective TIE2 inhibitors. This strategy can be adopted into general use for generating selective molecules with a desired pharmacophore.
Introduction
The aim of drug design is the discovery of new molecules with desirable pharmacological properties, which are beneficial for the treatment of diseases. To achieve the aim requires huge investment, resources, and a long time span from discovery to market.1,2) Drug design is still a complex and exceedingly difficult process in medicinal chemistry. It has been estimated that the size of drug-like chemical space is between 1023 to 1080 compounds.3) There have been many technologies utilized to discover novel candidate molecules, including high throughput screening, combinatorial libraries, and others.4–6) In the context of in silico drug discovery, various techniques, such as virtual screening and de novo drug design, have been proposed to discover novel molecular structures in the vast chemical space.7,8) Early works of de novo drug design used various algorithms, such as atom based elongation, fragment based combination, and reaction-driven design, to generate new molecular structures.9–13) The structure–activity relationship (SAR) Matrix (SARM) method was designed to combine SAR visualization with new compound design. It was originally introduced for the organization of related series of active compounds from data sets, visualization of SAR patterns, and generation of new compounds to further expand and optimize existing series.14) Most of the algorithms are based on the strategy of medicinal chemists: new molecules are synthesized (virtually generated using reaction data,11) etc.), evaluated with biological assays (evaluated by a scoring function9)), and optimized with medicinal chemistry (optimized with optimization algorithm, such as evolutionary algorithm12) and particle swarm optimization13)). Data-driven methods serve as robust tools in turning vast data in knowledge without introducing rule-based methods. Deep generative models (DGM) have been successfully applied to generate synthetic images,15) natural language texts,16) and others. DGM for the de novo design of molecular structures has gained popularity in recent years.17) Gómez-Bombarelli et al. adopted a variational autoencoder to generate molecular structures. Molecules represented as SMILES strings are encoded to real vectors in latent space. Novel SMILES strings are generated in the decoding process from real vectors to molecules.18) Adversarial autoencoder was employed to generate ligand against dopamine type 2 receptor.19) To generate drug-like molecules, satisfying multiple properties at the same time, conditional variational autoencoder was adopted.20) SMILES strings are generated from a graph representation of molecules. In GraphVAE, DGM is trained directly on the graph representation.21) Generative adversarial networks was adapted to DGM for small molecular graph, which is capable of generating close to 100% valid compounds in experiments on the QM9 chemical database.22) Deep reinforcement learning methods to generate molecules with desired properties have been proposed.23,24) In these methods, a pre-trained DGM is modified to generate molecules with the desired properties. Although DGMs are able to generate molecules with desired properties, most of the properties do not have three-dimensional (3D) information, such as shape and pharmacophore. It has been known that properties originating from 3D shape and/or pharmacophore are very useful in the drug design process, and very recently, shape-based generative model has been reported.25)
In this study we propose an approach to generate molecules with desired pharmacophore using deep reinforcement learning. Furthermore, generated molecules which has selectivity against a target protein are extracted using chemical genomics-based virtual screening (CGBVS) method.26,27)
Experimental
The methodology of molecular de novo design through deep reinforcement learning has been published elsewhere.23) The methodology has two recurrent neural network (RNN) models named as Prior and Agent networks. The Prior network is trained using SMILES strings from ChEMBL.28) After the training, the Prior network generates SMILES strings corresponding to valid molecular structures. The Agent network is trained using reinforcement learning. The training shifts the probability distribution from that of the Prior network towards a distribution modulated by a scoring function. The trained Agent network generates SMILES string with a desired property obtained from scoring function. The workflow of our strategy is shown in Fig. 1, consisting of three sections: Prior network construction, Agent network construction, and Structure sampling. In this study, Prior and Agent network construction were implemented using REINVENT.29) A pre-constructed Prior network was used. Training of the Agent network was done with a batch size of 64 using the Adam optimizer for 10000 steps. All other parameters were set to default values. LigandScout 4.330,31) was used as the scoring function. The pharmacophore model was constructed from the crystal structure of TIE2 and its inhibitor (PDB32) ID:2OSC) (Fig. 2). Here, eight pharmacophore features of the ligand were identified as two aromatic, two hydrogen acceptor, and four hydrophobic features. The four hydrophobic features are set as ‘optional feature.’ In addition, an ensemble of exclusion volume spheres obtained from the crystal structure was used. For pharmacophore scoring, scoring function is set to ‘Relative Pharmacophore-Fit’; maximum number of omitted features is set to ‘1’ for iscreen tool provided with LigandScout 4.3. the Relative Pharmacophore-Fit (rel.SFCR) was defined as Eq. 1.30)
 | (1) |
where SFCR is the feature count/RMS distance score, SRMS is the matched feature pair RMS distance score in the range [0,9], RMSFP is the RMS of the matched feature pair distances, and NMFP is the number of geometrically matched feature pairs. The parameter of conformer generation of generated structures from the Agent network is set as ‘iCon Fast33)’ option for idbgen tool provided with LigandScout 4.3. For similarity scoring, Tanimoto coefficient based on Morgan fingerprints (length = 2048, radius = 2, useCounts = True, useFeatures = True) using RDKit34) is used. The reference compound for the similarity scoring score is the TIE2 inhibitor illustrated in Fig. 2B. CGBVS26) score shows probabilities of protein-compound interactions. Herein, the CGBVS scores were calculated using CzeekS.27) For filtering of selective kinase inhibitors, 6000 valid SMILES strings (structures) were generated during structure sampling. CGBVS scores of the generated structures from the Agent and 412 kinases were calculated. The CGBVS scores were used for filtering selective kinase inhibitors. CGBVS scores range from 0 to 1. For a given compound, the selectivity score S(x),35) where x represents the threshold of CGBVS score, is calculated by dividing the number of kinases having a CGBVS score greater than x by the total number of kinases, as shown in Eq. 2.
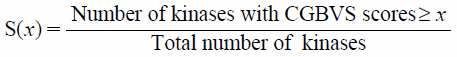 | (2) |
where CGBVS score against TIE2 must be greater than or equal to x and x is set to 0.9. A compound having smaller value for the selectivity score indicates a more selective TIE2 inhibition.
Results and Discussion
TIE2 kinase signaling pathway has a crucial role in both angiogenesis and tumorigenesis.36,37) It has been established that inhibition of the signaling pathway provide a potentially viable therapeutic strategy in cancer treatment.38) In this study, structures of selective TIE2 inhibitors were generated using deep reinforcement learning. The workflow of our strategy for identification of selective TIE2 inhibitors is shown in Fig. 1, which is described in detail in Experimental.
Pharmacophore ModelingThe crystal structure of TIE2 in complex with a pyrimidine derivative (PDB ID:2OSC) was selected for pharmacophore modeling (Figs. 2A, B). The pyrimidine derivative formed two hydrogen bond interactions with NH groups of the main chain of ALA905 and ASP982. The two aromatic rings of the pyrimidine derivative form hydrophobic interactions with the ATP binding pockets (ALA853, VAL838, PHE983, and LEU971) of TIE2. Furthermore, four hydrophobic features were derived from the crystal structure, which are defined as optional pharmacophore features. The methyl-benzyl group consisting of two hydrophobic features interacts with ILE902 and LEU900. The trifluoromethyl-benzyl group consisting of two hydrophobic features interacts with LEU876, VAL875, LEU879, ILE886, ALA981, LEU955, PHE960, and ILE980. In addition, an ensemble of exclusion volume spheres was obtained from the crystal structure to represent the shape of the active site. These features were defined as the pharmacophore in this study.
Comparison of Structures Generated from Prior and Agent NetworksAfter the training of the Agent network, 6000 structures are generated. In the same way, 6000 structures are generated from the Prior network. The percentages of valid SMILES generated by the Agent and Prior networks are 92.1% and 93.8%, respectively. The pharmacophore scores of the generated structures were calculated using LigandScout 4.3. The distribution of pharmacophore scores is displayed as a histogram in Fig. 3A. The 3763 structures having pharmacophore scores greater than 0.3 were confirmed during structure sampling from the Agent network. On the other hand, there are only 482 structures having pharmacophore scores greater than 0.3 during sampling from the Prior network. This result indicates that the Agent network can generate structures fulfilling the pharmacophore profile of a TIE2 inhibitor. Figure 3B shows the visual representation of the chemical space of structures generated by the Prior network (6000 structures) and the Agent network (1565 structures having pharmacophore scores greater than 0.5). The distribution of the structures generated from the Agent network is obviously different from the Prior network. From the results, we have confirmed that the Agent network could generate structures with the pharmacophore of a TIE2 inhibitor in a new region in the chemical space. To compare difference between the pharmacophore scoring based on the 3D information and a scoring based on the non-3D information, the Agent network trained by similarity scoring is constructed. The similarity score is calculated using Tanimoto coefficient based on Morgan fingerprints. The reference compound of the similarity score is the TIE2 inhibitor illustrated in Fig. 2B. After the training using similarity scoring of the Agent network, 6000 structures were generated. The percentage of valid SMILES generated by the Agent networks is 97.8%. The distribution of similarity scores is displayed as a histogram in Fig. 3C. The 1810 structures having similarity scores greater than 0.4 were confirmed during structure sampling from the Agent network trained by similarity scoring. On the other hand, there are only 114 and 195 structures having similarity scores greater than 0.4 during sampling from the Prior network and the Agent network trained by pharmacophore scoring, respectively. These results indicate that the Agent network trained by similarity scoring can generate structurally similar compound to the TIE2 reference compound. Interestingly, the Agent network trained by pharmacophore scoring can generate the compounds having higher similarity scores than that of Prior network. However, as illustrated in Fig. 3D, the Agent network trained by similarity scoring can hardly generate the compounds having pharmacophore scores greater than 0.1. The reason is that most of the generated compounds have contact with the exclusion volume spheres of the pharmacophore in this study.
Generated Structures with Desired PharmacophoreThe Agent network generated structures having the pharmacophore profile of a TIE2 inhibitor. A sample of the structures is shown in Fig. 4. The pyrimidine derivative has two aromatic rings (Ar1 and Ar2), two hydrophobic acceptors (Hac1, Hac2), and four hydrophobic features (Hp1, Hp2, Hp3, and Hp4). MOL03588, which has the highest pharmacophore score (0.75), has two aromatic rings (Ar1 and Ar2), two hydrogen bond acceptors (Hac1, Hac2), and two hydrophobic features (Hp1, Hp2). MOL03588 has an imidazole group as Hac1 feature instead of a pyrimidine group of the pyrimidine derivative. Hac1 feature shows interaction with ALA905 on the hinge region of TIE2 (Fig. 2). MOL00571 has two aromatic rings (Ar1 and Ar2), two hydrogen bond acceptors (Hac1, Hac2), and two hydrophobic features (Hp2, Hp3). MOL00571 has a triazole group as Hac1 feature. The Hac2 and Hp3 features are carbonyl and benzyl group, respectively (same as the pyrimidine derivative). MOL04081 has two aromatic rings (Ar1 and Ar2), two hydrogen bond acceptors (Hac1, Hac2), and two hydrophobic features (Hp2, Hp3). MOL04081 has pyridine groups as Hac1 and Hac2 feature, respectively. MOL04833 and MOL05367 has two aromatic rings (Ar1 and Ar2), two hydrogen bond acceptors (Hac1, Hac2), and one hydrophobic feature (Hp2). These molecules are quite different in structure compared with the pyrimidine derivative.
There were 1565 structures having pharmacophore scores greater than 0.5 confirmed during structure sampling from the Agent network. These structures were filtered using the selectivity scores obtained using Eq. 2. MOL03802 was identified as the most selective TIE2 inhibitor generated from the Agent network trained by pharmacophore scoring (Figs. 5A, B). CGBVS scores against 412 kinases are shown in Fig. 5C. CGBVS scores (> 0.8) of MOL03802 are 0.93 (TIE2), 0.89 (MK01), 0.83 (PIM1), and 0.80 (FRK). On the other hand, MOL03740FP was identified as the most selective TIE2 inhibitor generated from the Agent network trained by similarity scoring (Fig. 5D). CGBVS scores (> 0.8) of MOL03740FP are 0.94 (TIE2), 0.90 (MK09), 0.88 (FLT3), 0.87 (VGFR2), 0.86 (RAF1), 0.82 (TNI3K), 0.82 (CSF1R), 0.81 (BTK), and 0.81 (FGR), respectively (Fig. 5E). These results suggest that MOL03802 is a more selective TIE2 inhibitor than MOL03740FP. As illustrated in Fig. 5F, CGBVS scores of the reference TIE2 inhibitor show that the inhibitor is not so selective for TIE2. Therefore, compounds similar to the reference TIE2 inhibitor may not be selective inhibitors for TIE2.
Conclusion
This study proposes a strategy for design of molecular structures with a desired pharmacophore using deep reinforcement learning. The strategy has three sections consisting of Prior network construction, Agent network construction, and structure sampling. The Prior network is trained using SMILES strings from ChEMBL. After the training, the Prior network generates valid SMILES strings that include new molecular structures. The Agent network is trained using reinforcement learning. The training shifts the probability distribution from that of the Prior network towards a distribution modulated by a scoring function. Herein, pharmacophore scores obtained from LigandScout 4.3 are used as output of the scoring function. After the training, the Agent network generates valid SMILES strings that include new molecular structures having the pharmacophore profile of TIE2 inhibitors. About 26% of structures generated from the Agent network have the pharmacophore profile of TIE2 inhibitors and whose scores are greater than 0.5. This result shows that the Agent network learned the 3D pharmacophore information via deep reinforcement learning. The molecules generated from the Agent network should be useful novel seed compounds for further development of TIE2 inhibitors. Additionally, this study showed the utility of using CGBVS scores to extract structures of selective TIE2 inhibitors. Selectivity prediction is important in identifying potential compounds that inhibit the target specifically. The CzeekS kinase prediction model enables prediction of the probabilities of protein-compound interactions with respect to 412 kinases. Finally, MOL03802 is predicted as a selective TIE2 inhibitor. These results lead to the conclusion that our strategy can be a beneficial tool for generating selective structures with a desired pharmacophore against specific target proteins.
Acknowledgments
We would like to thank inte:Ligand GmbH for the advice regarding LigandScout and Dr. Hirofumi Nakano for insightful comments and suggestions.
Conflict of Interest
A.Y. is CEO of the Institute for Theoretical Medicine, Inc., E.K. and C.K. are employees of INTAGE Healthcare Inc., and T.T. is CEO of the Affinity Science Corporation. The authors declare no other potential conflict of interest.
References
- 1) DiMasi J. A., Grabowski H. G., Hansen R. W., J. Health Econ., 47, 20–33 (2016).
- 2) Paul S. M., Mytelka D. S., Dunwiddie C. T., Persinger C. C., Munos B. H., Lindborg S. R., Schacht A. L., Nat. Rev. Drug Discov., 9, 203–214 (2010).
- 3) Polishchuk P. G., Madzhidov T. I., Varnek A., J. Comput. Aided Mol. Des., 27, 675–679 (2013).
- 4) Macarron R., Banks M. N., Bojanic D., Burns D. J., Cirovic D. A., Garyantes T., Green D. V., Hertzberg R. P., Janzen W. P., Paslay J. W., Schopfer U., Sittampalam G. S., Nat. Rev. Drug Discov., 10, 188–195 (2011).
- 5) Liu R., Li X., Lam K. S., Curr. Opin. Chem. Biol., 38, 117–126 (2017).
- 6) Price A. J., Howard S., Cons B. D., Essays Biochem., 61, 475–484 (2017).
- 7) Rester U., Curr. Opin. Drug Discov. Devel., 1, 559–568 (2008).
- 8) Button A., Merk D., Hiss J. A., Schneider G., Nat. Mach. Intell., 1, 307–315 (2019).
- 9) Böhm H. J., J. Comput. Aided Mol. Des., 6, 61–78 (1992).
- 10) Mauser H., Stahl M., J. Chem. Inf. Model., 47, 318–324 (2007).
- 11) Hartenfeller M., Zettl H., Walter M., Rupp M., Reisen F., Proschak E., Weggen S., Stark H., Schneider G., PLOS Comput. Biol., 8, e1002380 (2012).
- 12) Devia R. V., Sathya S. S., Coumarb M. S., Appl. Soft Comput., 27, 543–552 (2015).
- 13) Czeek D., “De novo design system with PSO”: ‹ https://www.insilico.jp/czeekd.html›
- 14) Wassermann A. M., Haebel P., Weskamp N., Bajorath J., J. Chem. Inf. Model., 52, 1769–1776 (2012).
- 15) Goodfellow I. J., Pouget-Abadie J., Mirza M., Xu B., Warde-Farley D., Ozair S., Courville A., Bengio Y., arXiv:1406.2661 (2014).
- 16) Zhang Y., Gan Z., Fan K., Chen Z., Henao R., Shen D., Carin L., arXiv:1706.03850v3 (2017).
- 17) Elton D. C., Boukouvalas Z., Fuge M. D., Chung P. W., arXiv:1903.04388v3 (2019).
- 18) Gómez-Bombarelli R., Wei J. N., Duvenaud D., Hernández-Lobato J. M., Sánchez-Lengeling B., Sheberla D., Aguilera-Iparraguirre J., Hirzel T. D., Adams R. P., Aspuru-Guzik A., ACS Cent. Sci., 4, 268–276 (2018).
- 19) Blaschke T., Olivecrona M., Engkvist O., Bajorath J., Chen H., Mol Inform., 37, 1700123 (2018).
- 20) Lim J., Ryu S., Kim J. W., Kim W. Y., J. Cheminform., 10, 31 (2018).
- 21) Simonovsky M., Komodakis N., arXiv:1802.03480v1 (2018).
- 22) Cao N. D., Kipf T., arXiv:1805.11973v1 (2018).
- 23) Olivecrona M., Blaschke T., Engkvist O., Chen H., J. Cheminform., 9, 1 (2017).
- 24) Guimaraes G. L., Sanchez-Lengeling B., Outeiral C., Farias P. L. C., Aspuru-Guzik A., arXiv:1705.10843 (2017).
- 25) Skalic M., Jiménez J., Sabbadin D., De Fabritiis G., J. Chem. Inf. Model., 59, 1205–1214 (2019).
- 26) Yabuuchi H., Niijima S., Takematsu H., Ida T., Hirokawa T., Hara T., Ogawa T., Minowa Y., Tsujimoto G., Okuno Y., Mol. Syst. Biol., 7, 472 (2011).
- 27) Czeek S., “Screening system using Chemical Genomics-Based Virtual Screening (CGBVS)”: ‹https://www.insilico.jp/czeeks.html›
- 28) Bento A. P., Gaulton A., Hersey A., Bellis L. J., Chambers J., Davies M., Krüger F. A., Light Y., Mak L., McGlinchey S., Nowotka M., Papadatos G., Santos R., Overington J. P., Nucleic Acids Res., 42 (D1), D1083–D1090 (2014).
- 29) REINVENT, “Molecular de novo design using recurrent neural networks and reinforcement learning.”: ‹https://github.com/MarcusOlivecrona/REINVENT›
- 30) “Ligand Scout User Manual.”: ‹http://www.inteligand.com›
- 31) Wolber G., Dornhofer A. A., Langer T., J. Comput. Aided Mol. Des., 20, 773–788 (2007).
- 32) Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E., Nucleic Acids Res., 28, 235–242 (2000).
- 33) Poli G., Seidel T., Langer T., Front Chem., 6, 229 (2018).
- 34) RDKit, “Open-source cheminformatics.”: ‹http://www.rdkit.org›
- 35) Bosc N., Meyer C., Bonnet P., BMC Bioinformatics, 18, 17 (2017).
- 36) Saharinen P., Eklund L., Pulkki K., Bono P., Alitalo K., Trends Mol. Med., 17, 347–362 (2011).
- 37) Rigamonti N., Kadioglu E., Keklikoglou I., Wyser Rmili C., Leow C. C., De Palma M., Cell Reports, 8, 696–706 (2014).
- 38) Harney A. S., Karagiannis G. S., Pignatelli J., Smith B. D., Kadioglu E., Wise S. C., Hood M. M., Kaufman M. D., Leary C. B., Lu W. P., Al-Ani G., Chen X., Entenberg D., Oktay M. H., Wang Y., Chun L., De Palma M., Jones J. G., Flynn D. L., Condeelis J. S., Mol. Cancer Ther., 16, 2486–2501 (2017).




