Experimental
ChemistryMelting points were determined in capillary tubes on an electrothermal PIF YRT-3 apparatus and without correction. IR spectra were determined on NEXUS 670 FT-IR (Nicolet). 1H-NMR and 13C-NMR (δ ppm) spectra were recorded on a Varian Mercury (400 MHz) using tetramethylsilane (TMS) as the internal standard. Mass spectra were recorded on a VGZAB-HS (70 eV) spectrometer with electrospray ionization (ESI) source as ionization. All reactions were monitored by analytical TLC and silica gel F254 was used in TLC. All chemicals were purchased from commercial suppliers and were dried and purified when necessary.
General Synthetic Procedure for the Key Intermediates 2-Acetoxy Benzoyl Chloride (c7)46)Under anhydrous conditions, pyridine as the catalyst,47) higher yield of product can be obtained without any solvent. Thionyl chloride (4.00 mL) was added dropwise to a solution of ASA (0.05 mol) in the pyridine (0.02 mL) below 50°C. The suspension was gradually heated to 90°C, stirred and refluxed for 3h. The excess thionyl chloride was removed at 40°C under reduced pressure. The white viscous substance c7 (9.43g, 90%) was sealed in the desiccator.
General Procedure for the Synthesis of 1a38–40) A: Preparation of 2-Acetoxy Benzoyl Pyrrolidone (1.3a)48)Benzyltriethylaminium chloride (TEBA) (0.005 mol) was added to the pyrrolidone sodium salt (1.2a) (0.05 mol) dissolved in the anhydrous toluene solution below 10°C and c7 (0.05 mol) in anhydrous toluene was added dropwise. Then reacted at room temperature for 0.5 h and at 50°C for 3 h. Water (100 mL) was added and saturated sodium bicarbonate was mixed to wash the toluene layer with 20 mL, 20 mL, 10 mL, respectively, and then the suspension was washed until neutral with water (50 mL), dried over anhydrous magnesium sulfate overnight and crystallized with ethanol to precipitate the product 10.1 g, 81.7%.
B: Perparation of 1aPtO2 (12.5 mmol) was added to 2-acetoxy benzoyl pyrrolidone (25 mmol) dissolved in HCl (80 mL) and then the suspension was refluxed 12 h at 60°C with a yield of 72%. mp: 101.2–102.2°C. Rf 0.38 (eluent : petroleum : ethyl acetate : glacial acetic acid=3 : 2 : 0.02) IR (KBr) ν (cm−1): 3381, 3087, 1738, 1582–1445, 1241–1180; 1H-NMR (400 MHz, CDCl3) δ: 1.95 (m, 2H, J=6.6 Hz, J=6.9 Hz), 2.15 (s, 3H), 2.29 (t, 2H, J=5.4 Hz), 3.48 (m, 2H, J=6.0 Hz, J=6.6 Hz), 6.77–6.96 (m, 2H, J=7.5 Hz, J=8.1 Hz), 7.21–7.38 (m, 2H, J=3.9 Hz, J=9.0 Hz), 9.92 (br s, 1H); EI-MS (m/z): 222 [M+−43], 220 [M+−45], 178 [M+−87], 120 [M+−145]
General Synthetic Procedure for the Key Intermediates γ-Aminobutyrate p-Toluenesulfonate Salt (a2)43,44)γ-Aminobutyrate p-toluenesulfonate salt was prepared in a manner similar to that of Shields, McGregor, and Carpenter. GABA (20 mmol), benzenesulfonic acid monohydrate (22.88 mmol), benzyl alcohol (25 mmol) and benzene (25 mL) were refluxed overnight using a Dean–Stark apparatus for the removal of water. The solvent was then reduced to half its original volume under reduced pressure. Anhydrous ether was added after which precipitation occurred.
General Procedure for the Synthesis of 1a–8a43,44)The prepared c7 (22 mmol) dissolved in CH2Cl2 (8 mL) was added to a2 (20 mmol) mixed with CH2Cl2 60 mL dropwise and triethylamine (15 mL) was added to the suspension, which was placed in a reflux bath 60°C for 18–24 h to generate a1.
Triethylamine (5 mL) was added to a1 (10.6 mmol) dropwise dissolved in CH2Cl2 (50 mL) by stirring in ice-water bath for 10 min. Acetyl chloride (21.2 mmol) dissolved in CH2Cl2 (5 mL) was added to the suspension dropwise. The reaction was gradually restored to the room temperature after removing the ice-water bath. After completion of the reaction, water (20 mL) was added to quench reactions and the organic layer was washed with water (2×30 mL), dried with anhydrous sodium sulfate overnight. Removing the CH2Cl2 under reduced pressure, we can get 1a–8a.
General Procedure for the Synthesis of 1b–7b43,44) A: The Esterification of GABATo an ice-cold solution of GABA (1 equiv) and alcohol (1 equiv) in tetrahydrofuran (THF) (60 mL), SOCl2 (1.5 equiv) was added dropwise, and then the mixture was stirred at room temperature for 12h. The resulting slurry was evaporated and EtOAc was added. The mixture was stirred at room temperature and triethylamine (TEA) was added dropwise, filtered and evaporated to give a residual yellowish oil. The crude product was chromatographed (n-hexane : EtOAc=5 : 1) to give intermediates b1.
B: Procedure B Using Dicyclohexylcarbodiimide (DCC)An ice-cold solution of b1 (1 equiv), ASA (1.1 equiv) and 4-dimethyl aminopyridine (DMAP) (0.3 equiv) in CH2Cl2 (60 mL) was added with DCC (1.1 equiv) portionwise and the mixture was stirred at room temperature for 16 h. The resulting slurry (precipitated dicyclohexyl urea, DCU) was ice-cooled for 2 h and filtered. The collected solid was washed with ice-cold CH2Cl2, the filtrate was evaporated, and the residue was redissolved in EtOAc (70 mL), cooled, and filtered again (to precipitate any remaining DCU). The EtOAc solution was then washed with 5% citric acid solution, NaHCO3 solution, and finally brine. The solution was then concentrated under vacuum at 40°C to yield the final product as viscous orange oil.
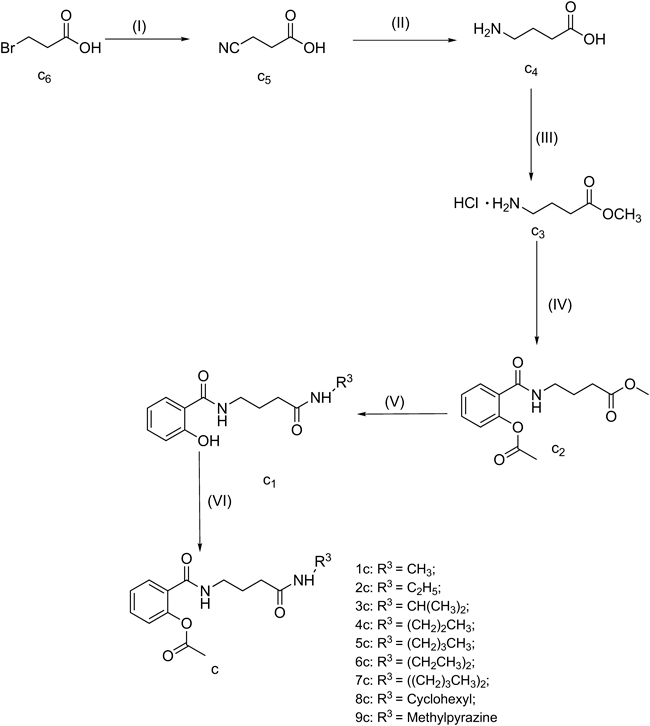
General Procedure for the Synthesis of 1c–9cDuring the ammonolysis of chloride, carboxyl group in GABA tended to cause cyclization and formation of the mixed anhydride. Generally a procedure easy to be stripped off were needed for its protection. Referred to the literature,44–49) the following synthetic route was designed.
A: Synthesis of c350–51)To an ice-cold solution of GABA (1 equiv) in methanol (50 mL), SOCl2 (1.2 equiv) was added dropwise and then the mixture was heated to reflux for 1h to yield the white solid in vacuum at 40°C. The product was recrystallized in ethanol–ether (1 : 2) solution. Yield 94%, mp 118.3–120°C.
B: Synthesis of c252,53)A mixture of c3 (23.00 mmol) and TEA (5 mL) in CH2Cl2 was added to c7, dissolved in CH2Cl2 by dropwise and then the mixture was heated to reflux for 2h and partitioned between CH2Cl2 and water. The aqueous phase was extracted with CH2Cl2. The combined organic layer was washed with NaHCO3 and water, dried, filtered, and evaporated to give yellowish oil (6.10g). Yield 85.3%.
C: Synthesis of 1c–9c54–56)A mixture of c2 (6.1 g) and amine (10 mL) in methanol (50 mL) was stirred at room temperature for 36 h, and then the mixture was vacuumed at 40°C and partitioned between CH2Cl2 and water. The organic layer was washed with NaHCO3 and water, dried (MgSO4), filtered, and evaporated to give yellowish oil with the yield of 80% (c1). Then c1 and TEA was dissolved in CH2Cl2 which was cooled to 0°C and chloride (3.0 mL), dissolved in CH2Cl2, was added by dropwise. The reaction was monitored by TLC (n-hexane : EtOAc=1 : 2). The water was added, partitioned between CH2Cl2 and water. The organic layer was washed with water, dried (MgSO4), filtered, and evaporated to give yellowish oil.
4-(2-Acetoxybenzoylamino)butyric Acid Ethyl Ester (2a)1.693 g, Yield, 54.5%; IR (KBr) ν (cm−1): 3326, 1735, 1645; 1H-NMR (400 MHz, CDCl3) δ: 1.22 (t, 3H, J=6.9 Hz), 1.92 (m, 2H, J=6.6 Hz, J=6.9 Hz), 2.13 (s, 3H), 2.25 (s, 3H), 2.42 (t, 2H, J=6.9 Hz), 3.47 (m, 2H, J=6.6 Hz), 4.09 (q, 2H, J=6.9 Hz), 6.77–7.53 (m, 4H, J=1.8 Hz, J=6.9 Hz, J=8.4 Hz), 7.15 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 13.8 (–CH2–CH3), 21.6 (–CH3), 23.9 (–CH2–CH2–CH2–), 31.6 (–CH2–CH2–CH2–), 39.0 (–CH2–CH2–CH2–), 60.4 (–CH2–CH3), 114.2, 117.9, 118.4, 119.5, 125.7, 133.7 (Ar), 160.9 (–NH–C=O), 169.9 (CH3–C=O), 173.6 (–CH2–C(=O)–O); EI-MS (m/z): 293[M]+, 294 [M+1]+, 121[M+−172].
4-(2-Acetoxybenzoylamino)butyric Acid Iospropyl Ester (3a)1.562 g, Yield 47.9%; IR (KBr) ν (cm−1): 3376, 1703, 1643; 1H-NMR (400 MHz, CDCl3) δ: 1.23 (d, 6H, J=6.4 Hz), 1.96 (t, 2H, J=6.4 Hz), 2.17 (s, 3H), 2.44 (m, 2H, J=6.4 Hz, J=6.8 Hz), 3.50 (t, 2H, J=6.8 Hz), 5.01 (m, 1H, J=6.4 Hz), 6.84–7.44 (m, 4H, J=1.2 Hz, J=2.0 Hz, J=5.2 Hz, J=7.2 Hz), 7.35 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 21.7 (–CH3), 23.8 (–CH–(CH3)2, 28.4 (–CH2–CH2–CH2–), 32.5 (–CH2–CH2–CH2–C=O), 39.6 (–NH–CH2–CH2–CH2–), 68.3 (–CH– (CH3)2), 114.2, 118.4, 118.6, 125.5, 132.3, 134.0 (Ar), 161.5 (–NH–C=O), 170.1 (CH3–C=O), 173.7 (–CH2–C(=O)); EI-MS (m/z): 307[M]+, 308[M+1]+, 121 [M+−172].
4-(2-Acetoxybenzoylamino)butanoic Acid n-Butyl Ester (4a)1.627 g, Yield 47.8%; IR (KBr) ν (cm−1): 3386, 1713, 1659; 1H-NMR (400 MHz, CDCl3) δ: 0.90 (t, 3H, J=7.2 Hz), 1.33 (m, 2H, J=5.2 Hz, J=7.2 Hz), 1.58 (m, 2H, J=5.2 Hz, J=6.4 Hz), 1.95 (q, 2H, J=6.4 Hz, J=6.8 Hz), 2.11 (s, 3H), 2.45 (t, 2H, J=6.8 Hz), 3.49 (q, 2H, J=6.4 Hz), 4.06 (t, 1H, J=6.4 Hz), 6.80–7.48 (m, 4H, J=0.8 Hz, J=1.6 Hz, J=6.8 Hz, J=8.4 Hz), 7.40 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 13.0 (–(CH2)3–CH3), 20.4 (–CH2–CH2–CH2–CH3), 22.7 (O=C–CH3), 23.8 (–CH2–CH2–CH2–C=O), 31.4 (–CH2–CH2–CH2–CH3), 33.9 (–CH2–CH2–CH2–C=O), 40.8 (–NH–CH2–CH2–CH2–), 63.6 (–O–CH2–CH2–CH2–CH3), 114.1, 118.5, 118.8, 126.8, 133.6, 137.3 (Ar), 167.9 (–NH–C=O), 170.3 (CH3–C=O), 173.6 (–CH2–C(=O)–O); EI-MS (m/z): 321 [M]+, 294 [M+1]+, 121 [M+−172].
4-(2-Acetoxybenzoylamino)butyric Acid Cyclohexyl Ester (5a)1.635 g, Yield 44.4%; IR (KBr) ν (cm−1): 3378, 1709, 1641; 1H-NMR (400 MHz, CDCl3) δ: 1.26–1.52 (m), 1.70 (t), 1.80 (m, 2H), 2.23 (s, 3H), 2.42 (t, 2H), 3.28 (t, 2H), 4.73 (t), 6.84–7.42 (m, 4H, J=7.2 Hz), 7.27 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 23.7 (–O=C–CH3), 23.8 (C3, C5 of cyclohexanyl), 25.2 (C4 of cyclohexanyl), 29.7 (–CH2–CH2–CH2–), 31.5 (C2, C6 of cyclohexanyl), 32.6 (O=C–CH2–CH2–CH2–), 39.6 (–NH–CH2–CH2–CH2–), 73.3 (C1 of cyclohexanyl), 114.2, 118.5, 118.6, 125.5, 133.3, 134.0 (Ar), 161.6 (–NH–C=O), 170.1 (CH3–C=O), 173.7 (–C(=O)-O); EI-MS (m/z): 347 [M]+, 348 [M+1]+, 121 [M+−172].
4-(2-Acetoxybenzoylamino)butyric Acid Benzyl Ester (6a)1.524 g, Yield 40.4%; IR (KBr) ν (cm−1): 3316, 1741, 1635; 1H-NMR (400 MHz, CDCl3) δ: 1.97 (m, 2H, J=6.4 Hz, J=6.8 Hz), 2.17 (s, 3H), 2.46 (t, 2H, J=6.8 Hz), 3.50 (q, 2H, J=6.4 Hz), 5.34 (s, 1H), 6.82–6.97 (m, 4H, J=7.2 Hz, J=8.0 Hz), 7.18–7.44 (m, 4H, J=7.6 Hz, J=8.0 Hz), 7.18 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 20.1 (–CH3), 23.9 (–CH2–CH2–CH2–), 32.2 (–CH2–CH2– CH2–NH–), 38.9 (O=C–CH2–CH2– CH2–), 68.8 (–CH2–ph), 128.9 (C2, C6 of ph), 129.8 (C3, C5 of ph), 118.6, 120.4, 120.6, 122.4, 125.7, 127.7, 133.9, 135.4 (Ar and C4 of ph), 161.6 (–NH–C=O), 169.9 (CH3–C=O), 173.0 (–CH2–C(=O)–O); EI-MS (m/z): 355 [M]+, 356 [M+1]+, 121 [M+−172].
4-(2-Acetoxybenzoylamino)butyric Acid p-Methoxy Benzyl Ester (7a)1.952 g, Yield 47.8%; IR (KBr) ν (cm−1): 3314, 1734, 1642; 1H-NMR (400 MHz, CDCl3) δ: 1.97 (m, 2H, J=6.0 Hz, J=7.2 Hz), 2.18 (s, 3H), 2.51 (t, 2H, J=7.2 Hz), 3.49 (q, 2H, J=6.0 Hz), 3.81, 5.12 (s, 2H), 6.81–7.04 (m, 4H, J=7.6 Hz, J=8.8 Hz), 7.26–7.40 (m, 4H, J=3.2 Hz, J=4.8 Hz, J=8.8 Hz), 7.30 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 22.3 (O=C–CH3), 23.9 (–CH2–CH2–CH2–), 31.7 (–CH2–CH2–CH2–C=O), 39.1 (–NH–CH2–CH2–CH2–), 54.9 (–OCH3), 60.5 (–CH2–Ar), 113.5, 113.6, 113.7, 114.2, 114.2, 117.9, 118.4, 125.8, 129.4, 133.7 (2Ar), 161.0 (–NH–C=O), 169.9 (CH3–C=O), 173.7 (–CH2–C(=O)); EI-MS (m/z): 385 [M]+, 386 [M+1]+, 121 [M+−172].
4-(2-Acetoxybenzoylamino)butyric Acid p-Chloro Benzyl Ester (8a)2.121 g, Yield 51.3%; IR (KBr) ν (cm−1): 3378, 1702, 1644; 1H-NMR (400 MHz, CDCl3) δ: 2.01 (m, 2H, J=6.0 Hz, J=6.4 Hz), 2.18 (s, 3H), 2.50 (q, 2H, J=6.4 Hz), 3.50 (q, 2H, J=6.0 Hz), 5.07 (s, 2H), 6.81–7.27 (m, 4H, J=8.0 Hz, J=8.8 Hz), 7.30–7.40 (m, 4H, J=1.2 Hz, J=2.4 Hz, J=7.2 Hz, J=7.6 Hz), 7.30 (br s, 1H); 13C-NMR(100 MHz, CDCl3) δ: 23.7 (–CH3), 29.6 (–CH2–CH2–CH2–), 32.1 (–CH2–CH2–CH2–C=O), 39.3 (–NH–CH2–CH2–CH2–), 65.8 (–CH2–Ar), 118.5, 119.6, 125.4, 126.1, 128.1, 128.5, 129.8, 134.1, 135.4, 138.4 (2Ar), 163.5 (–NH–C=O), 171.6 (CH3–C=O), 173.1 (–CH2–C(=O)–O); EI-MS (m/z): 390 [M]+, 391 [M+1]+, 121 [M+−172].
4–(2-Acetoxybenzoylamino)butyric Acid Methyl Ester (1b)6.2 g, Yield, 87%; IR (KBr) ν (cm−1): 3328, 1748, 1653; 1H-NMR (400 MHz,CDCl3) δ: 1.87 (m, 2H, J=6.6 Hz, J=7.2 Hz), 2.33 (s, 3H), 2.41 (t, 2H, J=7.2 Hz), 3.44 (m, 2H, J=6.6 Hz, J=7.2 Hz), 3.68 (s), 7.08 (d, 1H, J4′–5′=7.1 Hz), 7.25 (m, 1H, J3′–4′=7.3 Hz, J2′–3′=6.4 Hz), 7.45 (m, 1H, J2′–4′=1.3 Hz, J3′–4′=7.3 Hz, J4′–5′=7.1 Hz), 7.66 (dd, 1H, J2′–4′=1.3 Hz, J2′–3′=6.4 Hz), 7.89 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 20.7 (CH3–C=O), 24.5 (–CH2–CH2–CH2–), 31.3 (–CH2–CH2–CH2–), 38.7 (–CH2–CH2–CH2–), 51.8 (–C(=O)–O–CH3), 122.8, 125.9, 128.3, 129.2, 131.6, 147.7 (6C, Ar), 165.7 (–NH–C=O), 169.5 (CH3–C=O), 173.7 (–CH2–C(=O)–O); RMS (ESI): (M+H+) 280.1189 (calculated 279.1107), error=2.3 ppm.
4-(2-Acetoxybenzoylamino)butyric Acid n-Propyl Ester (2b)5.8 g, Yield 84%; IR (KBr) ν (cm−1): 3338, 1757, 1648; 1H-NMR (400 MHz, CDCl3) δ: 1.86 (m, 2H, J=6.5 Hz, J=7.1 Hz), 2.38 (t, 2H, J=7.2 Hz), 3.44 (t, 2H, J=6.6 Hz, J=7.2 Hz,), 0.89 (t, J=6.2 Hz), 1.34 (q, J=6.2 Hz, J=6.8 Hz), 3.87 (t, J=6.8 Hz), 2.31 (s, 3H), 7.12 (d, 1H, J4′–5′=7.5 Hz), 7.24 (m, 1H, J3′–4′=7.8 Hz, J2′–3′=6.8 Hz), 7.43 (m, 1H, J2′–4′=1.2 Hz, J3′–4′=7.8 Hz, J4′–5′=7.5 Hz), 7.66 (dd, 1H, J2′–4′=1.2 Hz, J2′–3′=6.8 Hz), 7.86 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 21.2 (CH3–C=O), 24.8 (–CH2–CH2–CH2–C=O), 31.5 (–CH2–CH2–CH2–C=O), 37.9 (–CH2–CH2–CH2–C=O), 57.8 (–O–CH2–CH2CH3), 23.6 (–O–CH2–CH2–CH3), 11.5 (–O–CH2–CH2–CH3), 122.6, 125.7, 128.3, 129.1, 131.6, 147.9 (6C,Ar), 165.8 (–NH–C=O), 168.9 (CH3–C=O), 173.7 (–CH2–C(=O)–O); RMS(ESI): (M+NH4+) 325.1417 (calculated 307.1420), error=2.1 ppm.
4-(2-Acetoxybenzoylamino)butyric Acid 2,6-Dimethyl Phenyl Ester (3b)4.6 g, Yield 56%; IR (KBr) ν (cm−1): 3418, 1727, 1687; 1H-NMR (400 MHz, CDCl3) δ: 1.85 (m, 2H, J=6.1 Hz, J=7.6 Hz), 2.33 (t, 2H, J=7.6 Hz), 3.34 (t, 2H, J=6.1 Hz), 2.35 (s, 6H), 2.09 (s, 3H), 6.98–7.08 (m, 3H, J=5.4 Hz, J=7.2 Hz), 7.24–7.52 (m, 4H, J=2.2 Hz, J=5.6 Hz, J=6.8 Hz), 8.01 (s); 13C-NMR (100 MHz,CDCl3) δ: 21.6 (CH3–C=O), 23.8 (–CH2–CH2–CH2–C=O), 32.3 (–CH2–CH2–CH2–C=O), 39.1 (–CH2–CH2–CH2–C=O), 24.5 (2C, Ar–CH3), 119.3, 122.3, 123.6, 126.4, 127.1, 127.8, 128.5, 129.2, 131.5, 132.5, 145.8 (12C, 2Ar), 161.5 (–NH–C=O), 168.7 (CH3–C=O), 171.8 (–CH2–C=O); RMS(ESI): (M+NH4+) 387.1924 (calculated 369.1576), error=2.6 ppm.
4-(2-Acetoxybenzoylamino)butyric Acid 1-Phenyl Ethyl Ester (4b)4.6 g, Yield 56%; IR (KBr) ν (cm−1): 3363, 1682, 1716; 1H-NMR (400 MHz, CDCl3) δ: 1.88 (m, 2H, J=6.4 Hz, J=7.2 Hz), 2.33 (t, 2H, J=7.2 Hz), 3.34 (t, 2H, J=6.4 Hz), 2.13 (s, 3H), 1.67 (d, 3H, J=6.4 Hz), 4.84 (q, 1H, J=6.4 Hz), 6.78–7.22 (m, 5H, J=1.2 Hz, J=5.6 Hz, J=7.6 Hz) 7.24–7.51 (m, 4H, J=1.5 Hz, J=6.4 Hz, J=7.2 Hz); 13C-NMR (100 MHz, CDCl3) δ: 19.2 (-CH–CH3), 73.8 (–CH–CH3), 21.8 (CH3–C=O), 23.6 (–CH2–CH2–CH2–C=O), 32.2 (–CH2–CH2–CH2–C=O), 38.9 (–CH2–CH2–CH2–C=O), 24.3 (2C,Ar–CH3), 121.2, 122.0, 123.1, 126.1, 128.3, 128.5, 129.2, 131.5, 131.9, 147.9 (12C, 2Ar), 164.0 (–NH–C=O), 169.4 (CH3–C=O), 171.3 (–CH2–C=O); RMS(ESI): (M+NH4+) 387.2244 (calculated 369.1576).
4-(2-Acetoxybenzoylamino)butyric Acid o-Tolyl Ester (5b)4.9 g, Yield 60%; IR (KBr) ν (cm−1): 3313, 1719, 1646; 1H-NMR (400 MHz, CDCl3) δ: 1.85 (m, 2H, J=6.3 Hz, J=7.2 Hz), 2.33 (t, 2H, J=7.2 Hz), 3.37 (t, 2H, J=6.3 Hz), 2.36 (s, 3H), 2.18 (s, 3H), 6.83–7.05 (m, 4H, J=7.2 Hz, J=7.8 Hz), 7.26–7.40 (m, 4H, J=2.5 Hz, J=6.1 Hz, J=7.6 Hz), 7.91 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 21.3 (CH3–C=O), 25.8 (–CH2–CH2–CH2–C=O), 30.5 (–CH2–CH2–CH2–C=O), 41.4 (–CH2–CH2–CH2–C=O), 15.3 (Ar–CH3), 120.9, 122.0, 122.6, 126.1, 128.3, 129.2, 131.8, 132.3, 147.8 (2Ar), 164.1 (–NH–C=O), 168.6 (CH3–C=O), 172.1 (–CH2–C=O); RMS(ESI): (M+NH4+) 373.1913 (calculated 355.1420), error=2.1 ppm.
4-(2-Acetoxybenzoylamino)butyric Acid n-Amyl Ester (6b)4.7 g, Yield 62%; IR (KBr) ν (cm−1): 3411, 1736, 1640; 1H-NMR (400 MHz, CDCl3) δ: 1.82 (m, 2H, J=6.6 Hz, J=7.5 Hz), 2.38 (t, 2H, J=7.5 Hz), 3.44 (t, 2H, J=6.6 Hz), 0.87 (t, 3H, J=6.2 Hz), 1.21 (m, 2H, J=6.2 Hz, J=6.4 Hz), 1.37 (m, 2H, J=6.4 Hz, J=6.7 Hz), 1.24 (m, 3H, J=6.5 Hz, J=6.7 Hz), 3.95 (t, 3H, J=6.5 Hz), 2.28 (s, 3H), 7.12 (d, 1H, J4′–5′=7.2 Hz), 7.21 (m, 1H, J3′–4′=7.4 Hz, J2′–3′=6.6 Hz), 7.38 (m, 1H, J2′–4′=1.3 Hz, J3′–4′=7.4 Hz, J4′–5′=7.2 Hz), 7.51 (dd, 1H, J2′–4′=1.3 Hz, J2′–3′=6.6 Hz), 7.91 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 21.7 (CH3–C=O), 23.9 (–CH2–CH2–CH2–C=O), 32.3 (–CH2–CH2–CH2–C=O), 38.7 (–CH2–CH2–CH2–C=O), 12.4 (–O–CH2–CH2– CH2–CH2CH3), 22.3 (–O–CH2–CH2–CH2–CH2CH3), 25.9 (–O–CH2–CH2–CH2–CH2CH3), 29.2 (–O–CH2–CH2–CH2–CH2–CH3), 64.1 (–O–CH2–CH2– CH2–CH2CH3), 122.6, 126.3, 128.3, 129.2, 131.2, 147.7 (6C, Ar), 165.6 (–NH–C=O), 169.2 (CH3–C=O), 172.3 (–CH2–C(=O)–O); RMS(ESI): (M+NH4+) 353.1917 (calculated 335.1733), error=1.9 ppm.
4-(2-Acetoxybenzoylamino)butyric Acid iso-Amyl Ester (7b)4.9 g, Yield 63%; IR (KBr) ν (cm−1): 3342, 1737, 1671; 1H-NMR (400 MHz, CDCl3) δ: 1.85 (m, 2H, J=6.3 Hz, J=7.2 Hz), 2.33 (t, 2H, J=7.2 Hz), 3.37 (t, 2H, J=6.3 Hz), δ: 0.89 (d, 6H, J=6.2 Hz), 1.73 (m, 1H, J=6.2 Hz, J=6.8 Hz), 1.46 (m, 2H, J=6.8 Hz, J=6.3 Hz), 3.93 (t, 2H, J=6.3 Hz), 2.09 (s, 3H), 7.05 (d, 1H, J4′–5′=6.8 Hz), 7.19 (m, 1H, J3′–4′=7.2 Hz, J2′–3′=6.5 Hz), 7.35 (m, 1H, J2′–4′=1.5 Hz, J3′–4′=7.2 Hz, J4′–5′=7.2 Hz), 7.63 (dd, 1H, J2′–4′=1.5 Hz, J2′–3′=6.5 Hz), 7.93 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 22.3 (CH3–C=O), 24.6 (–CH2–CH2–CH2–C=O), 32.1 (–CH2–CH2–CH2–C=O), 38.6 (–CH2–CH2–CH2–C=O), 21.7 (2C,–CH(CH3)2), 23.7 (–CH(CH3)2), 33.7 (–O–CH2–CH2–), 61.9 (–O–CH2–CH2–), 122.2, 124.8, 127.7, 127.9, 132.5, 149.3 (6C, Ar), 164.3 (–NH–C=O), 168.3 (CH3–C=O), 175.5 (–CH2–C(=O)–O); RMS(ESI): (M+NH4+) 353.1712 (calculated 335.1733), error=2.1 ppm.
2-(3-(Methylcarbamoyl)propylcarbamoyl)phenyl Acetate (1c)4.74 g, Yield 63.5%. IR (KBr) ν (cm−1): 3281, 1721, 1648; 1H-NMR (400 MHz, CDCl3) δ: 1.87 (m, 2H, J=6.8 Hz, J=7.4 Hz), 2.30 (s, 3H), 2.39 (t, 2H, J=7.4 Hz), 2.76 (d, 3H, J=2.8 Hz), 3.40 (m, 2H, J=6.8 Hz), 7.07 (d, 1H, J4′–5′=6.6 Hz), 7.25 (m, 1H, J2′–3′=6.4 Hz, J3′–4′=6.2 Hz), 7.43 (m, 1H, J2′–4′=1.8 Hz, J3′–4′=6.4 Hz, J4′–5′=6.6 Hz), 7.65 (dd, 1H, J2′–4′=1.8 Hz, J2′–3′=6.2 Hz), 7.96–7.98 (br s, 2H); 13C-NMR (100 MHz, CDCl3) δ: 20.9 (1C, CH3–C=O), 24.5 (1C, –CH2–CH2–CH2–), 26.2 (1C, –CH3), 31.3 (1C, –CH2–CH2–CH2–), 39.5 (1C, –CH2–CH2–CH2–), 122.9, 126.2, 128.4, 129.2, 131.5, 147.9 (6C, Ar), 165.9 (1C, –NH–C=O), 169.2 (1C, CH3–C=O), 173.6 (1C, –CH2–C(=O)–O); HR-MS(ESI): (M+H+) 279.1391 (calculated 279.138), error=2.1 ppm.
2-(3-(Ethylcarbamoyl)propylcarbamoyl)phenyl Acetate (2c)4.86 g, Yield 67.2%; IR (KBr) ν (cm−1): 3357, 1768, 1734, 1659; 1H-NMR (400 MHz, CDCl3) δ: 1.25 (t, 3H, J=7.2 Hz), 1.87 (m, 2H, J=6.8 Hz, J=7.4 Hz), 2.30 (s, 3H), 2.39 (t, 2H, J=7.4 Hz), 3.39 (m, 2H, J=6.8 Hz), 3.53 (m, 2H, J=7.2 Hz), 7.17 (d, 1H, J4′–5′=6.6 Hz), 7.25 (m, 1H, J2′–3′=6.2 Hz, J3′–4′=6.4 Hz), 7.43 (m, 1H, J2′–4′=2.0 Hz, J3′–4′=6.4 Hz, J4′–5′=6.6 Hz), 7.65 (dd, 1H, J2′–4′=2.0 Hz, J2′–3′=6.2 Hz), 7.86–7.98 (br s, 2H); 13C-NMR (100 MHz, CDCl3) δ: 15.5 (1C, –CH2–CH3), 20.7 (1C, CH3–C=O) 24.2 (1C, –CH2–CH2–CH2–), 31.2 (1C, –CH2–CH2–CH2–), 34.2 (1C, –CH2–CH3), 39.5 (1C, –CH2–CH2–CH2–), 123.6, 126.6, 128.6, 129.5, 131.9, 145.8 (6C, Ar), 167.8 (1C, –NH–C=O), 169.1 (1C, CH3–C=O, 172.3 (1C,–CH2–C(=O)–O); HR-MS(ESI): (M+H+) 293.1523 (calculated 293.1517), error=2.0 ppm.
2-(3-(Isopropylcarbamoyl)propylcarbamoyl)phenyl Acetate (3c)5.13 g, Yield 61.4%; IR (KBr) ν (cm−1): 3311, 1767, 1734, 1646; 1H-NMR (400 MHz, CDCl3) δ: 1.28 (d, 6H, J=6.8 Hz), 1.89 (m, 2H, J=6.8 Hz, J=7.4 Hz), 2.33 (s, 3H), 2.43 (t, 2H, J=7.4 Hz), 3.33 (m, 2H, J=6.8 Hz), 3.88 (m, 1H, J=6.8 Hz), 7.01 (d, 1H, J4′–5′=6.8 Hz), 7.25 (m, 1H, J2′–3′=5.2 Hz, J3′–4′=6.4 Hz), 7.45 (m, 1H, J2′–4′=1.2 Hz, J3′–4′=6.4 Hz, J4′–5′=6.8 Hz), 7.64 (dd, 1H, J2′–4′=1.2 Hz, J2′–3′=5.2 Hz), 7.87–7.98 (br s, 2H); 13C-NMR (100 MHz, CDCl3) δ: 20.2 (1C, CH3–C=O), 23.7 (2C, –CH–(CH3)2), 26.3 (1C, –CH2–CH2–CH2–), 32.5 (1C, –CH2–CH2–CH2–), 39.8 (1C, –CH2–CH2–CH2–), 42.1 (1C, –CH–(CH3)2), 121.1, 124.2, 126.0, 127.2, 129.5, 146.1 (6C, Ar), 164.2 (1C, –NH–C=O), 168.2 (1C, CH3–C=O), 172.1 (1C, –CH2–C(=O)–O); HR-MS(ESI): M+H+) 307.1655 (calculated 307.1652), error=1.0 ppm.
2-(3-(Propylcarbamoyl)propylcarbamoyl)phenyl Acetate (4c)5.02 g, Yield 59.3%; IR (KBr) ν (cm−1): 3348, 1773, 1734, 1647; 1H-NMR (400 MHz, CDCl3) δ: 0.96 (t, 3H, J=7.2 Hz), 1.61 (m, 2H, J=7.2 Hz), 1.91 (m, 2H, J=6.8 Hz, J=7.2 Hz), 2.23 (s, 3H), 2.40 (t, 2H, J=6.8 Hz), 3.40 (m, 2H, J=7.2 Hz), 3.45 (m, 2H, J=7.2 Hz), 6.98 (d, 1H, J4′–5′=6.8 Hz), 7.25 (m, 1H, J2′–3′=6.4 Hz, J3′–4′=6.8 Hz), 7.43 (m, 1H, J2′–4′=2.0 Hz, J3′–4′=6.8 Hz, J4′–5′=6.8 Hz), 7.69 (dd, 1H, J2′–4′=2.0 Hz, J2′–3′=6.4 Hz), 7.96–8.02 (br s, 2H); 13C-NMR (100 MHz, CDCl3) δ: 11.2 (1C, –CH2–CH2–CH3), 20.6 (1C, CH3–C=O), 23.9 (1C, –CH2–CH2–CH2–), 25.9 (1C, –CH2–CH2–CH2–), 31.6 (1C, –CH2–CH2–CH2–), 39.8 (1C, –CH2–CH2–CH2–), 41.5 (1C, –CH2–CH2–CH3), 122.9, 126.3, 128.2 129.9, 131.5147.5 (6C, Ar), 165.6 (1C, –NH–C=O), 169.1 (1C, CH3–C=O), 173.1 (1C, –CH2–C(=O)–O); HR-MS(ESI): M+H+) 307.16 (calculated 307.1652), error=1.6 ppm.
2-(3-(Butylcarbamoyl)propylcarbamoyl)phenyl Acetate (5c)5.28 g, Yield 61.2%; IR (KBr) ν (cm−1): 3281, 1765, 1648; 1H-NMR (400 MHz, CDCl3) δ: 0.92 (t, 3H, J=6.0 Hz), 1.32 (m, 2H, J=6.0 Hz, J=6.2 Hz), 1.64 (m, 2H, J=5.8 Hz, J=6.2 Hz), 1.93 (q, 2H, J=6.4 Hz, J=7.2 Hz), 2.28 (s, 3H), 2.41 (t, 2H, J=6.4 Hz), 3.39 (q, 2H, J=7.2 Hz), 3.41 (t, 1H, J=5.8 Hz), 7.08 (d, 1H, J4′–5′=7.2 Hz), 7.26 (m, 1H, J2′–3′=6.4 Hz, J3′–4′=6.8 Hz), 7.44 (m, 1H, J2′–4′=1.6 Hz, J3′–4′=6.8 Hz, J4′–5′=7.2 Hz), 7.67 (dd, 1H, J2′–4′=1.6 Hz, J2′–3′=6.4 Hz), 7.86–7.92 (br s, 2H); 13C-NMR (100 MHz, CDCl3) δ: 13.5 (1C, –(CH2)3–CH3), 18.7 (1C, –CH2–CH2–CH2–CH3), 20.9 (1C, O=C–CH3), 26.5 (1C, –CH2–CH2–CH2–C=O), 31.1 (1C, –CH2–CH2–CH2–CH3), 33.6 (1C, –CH2–CH2–CH2–C=O), 39.5 (1C, –NH–CH2–CH2–CH2–), 41.5 (1C, –CH2–CH2–CH2–CH3), 122.5, 125.9, 127.6, 128.9, 131.3, 146.8 (6C, Ar), 166.1 (1C, –NH–C=O), 168.9 (1C, CH3–C=O), 173.1 (1C, –CH2–C(=O)–O); HR-MS(ESI): (M+H+) 321.1802 (calculated 321.1809), error=2.2 ppm.
2-(3-(Diethylcarbamoyl)propylcarbamoyl)phenyl Acetate (6c)4.95 g, Yield 58.3%. IR (KBr) ν (cm−1): 3356, 1768, 1733, 1660; 1H-NMR (400 MHz, CDCl3) δ: 1.11 (m, 6H, J=7.6 Hz), 1.81 (m, 2H, J=6.8 Hz), 2.16 (s, 3H), 2.40 (t, 2H, J=6.8 Hz), 3.39 (q, 2H, J=6.8 Hz), 3.42 (m, 4H, J=7.6 Hz), 7.10 (d, 1H, J4′–5′=6.4 Hz), 7.26 (m, 1H, J2′–3′=6.0 Hz, J3′–4′=6.4 Hz), 7.44 (m, 1H, J2′–4′=1.2 Hz, J3′–4′=6.4 Hz, J4′–5′=6.4 Hz), 7.67 (dd, 1H, J2′–4′=1.2 Hz, J2′–3′=6.0 Hz), 7.92 (br s, 2H); 13C-NMR (100 MHz, CDCl3) δ: 13.8 (2C, –CH2–CH3), 20.5 (1C,O=C–CH3), 26.9 (1C, –CH2–CH2–CH2–C=O), 31.3 (1C, –CH2–CH2–CH2–C=O), 39.2 (1C, –NH–CH2–CH2–CH2–), 42.0 (2C, –CH2–CH3), 122.3, 125.4, 126.1, 127.8, 131.4, 151.1 (6C, Ar), 167.5 (1C, –NH–C=O), 168.7 (1C, CH3–C=O), 171.5 (1C, –CH2–C(=O)–O); HR-MS(ESI): (M+H+) 321.1819 (calculated 321.1809), error=3.1 ppm.
2-(3-(Dibutylcarbamoyl)propylcarbamoyl)phenyl Acetate (7c)5.26 g, Yield 55.6%; IR (KBr) ν (cm−1): 3319, 1767, 1738, 1640; 1H-NMR (400 MHz, CDCl3) δ: 0.95 (t, 6H, J=7.2 Hz, J=7.6 Hz), 1.32 (m, 4H, J=7.2 Hz, J=7.6 Hz), 1.52 (m, 4H, J=4.6 Hz, J=7.6 Hz), 1.99 (m, 2H, J=6.4 Hz, J=7.2 Hz), 2.14 (s, 3H), 2.47 (t, 2H, J=6.4 Hz), 3.39 (q, 2H, J=7.2 Hz), 3.42 (t, 4H, J=4.6 Hz), 6.82 (d, 1H, J4′–5′=7.2 Hz), 6.87 (m, 1H, J2′–3′=6.4 Hz, J3′–4′=6.8 Hz), 7.47 (m, 1H, J2′–4′=1.0 Hz, J3′–4′=6.8 Hz, J4′–5′=7.2 Hz), 7.47 (dd, 1H, J2′–4′=1.0 Hz, J2′–3′=6.4 Hz), 7.83 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 13.7, 13.8 (2C, –CH2–CH2–CH2–CH3), 20.0, 20.1 (2C, –CH2–CH2–CH2–CH3), 21.4 (1C, O=C–CH3), 23.9 (1C, –CH2–CH2–CH2–C=O), 30.4, 30.9 (2C, –CH2–CH2–CH2–CH3), 31.7 (1C, –CH2–CH2–CH2–C=O), 39.3 (1C, –NH–CH2–CH2–CH2–), 48.0–48.1 (2C, –CH2–CH2–CH2–CH3), 114.3, 118.5, 125.8, 127.2, 129.7, 133.9 (6C, Ar), 161.4 (1C, –NH–C=O), 170.0 (1C, CH3–C=O), 174.4 (1C, –CH2–C(=O)–O); HR-MS(ESI): (M+H+) 377.2378 (calculated 377.2373), error=1.3 ppm.
2-(3-(Cyclohexylcarbamoyl)propylcarbamoyl)phenyl Acetate (8c)4.86 g, Yield 53.7%; IR (KBr) ν (cm−1): 3310, 1767, 1734, 1646; 1H-NMR (400 MHz, CDCl3) δ: 1.47 (m, 6H, J=6.8 Hz), 1.82 (m, 2H, J=6.4 Hz, J=7.2 Hz), 2.08 (s, 3H), 2.41 (t, 2H, J=6.4 Hz), 3.34 (q, 2H, J=7.2 Hz), 3.67 (t, 4H, J=6.8 Hz), 6.88 (d, 1H, J4′–5′=6.8 Hz), 7.05 (m, 1H, J2′–3′=6.4 Hz, J3′–4′=6.8 Hz), 7.20 (m, 1H, J2′–4′=1.6 Hz, J3′–4′=6.8 Hz, J4′–5′=6.8 Hz), 7.43 (dd, 1H, J2′–4′=1.6 Hz, J2′–3′=6.4 Hz), 7.78 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 20.1 (1CO=C–CH3), 25.2 (1C, –(CH2)2–CH2–(CH2)2–), 25.9 (1C, –CH2–CH2–CH2–C=O), 28.2–28.3 (2C, –CH2–CH2–CH2–CH2–CH2–), 31.7 (1C, –CH2–CH2–CH2–C=O), 6.9 (1C, –NH–CH2–CH2–CH2–), 41.8–41.9 (2C, –CH2–(CH2)3–CH2–), 114.8, 119.3, 125.8, 127.3, 129.6, 133.9 (6C, Ar), 161.3 (1C, –NH–C=O), 170.2 (1C, CH3–C=O), 173.9 (1C, –CH2–C(=O)–O). HR-MS(ESI): (M+H+) 333.1798 (calculated 333.1792), error=1.8 ppm.
2-(3-(Methylpyrazinecarbamoyl)propylcarbamoyl)phenyl Acetate (9c)4.57 g, Yield 51.6%; IR (KBr) ν (cm−1): 3318, 1770, 1737, 1638; 1H-NMR (400 MHz, CDCl3) δ: 1.85 (m, 2H, J=6.8 Hz, J=7.2 Hz), 2.09 (s, 3H), 2.35 (t, 2H, J=7.2 Hz), 2.53 (s, 3H), 2.74 (t, 4H, J=6.4 Hz), 3.25 (q, 2H, J=6.8 Hz), 3.51 (t, 4H, J=6.4 Hz), 6.97 (d, 1H, J4′–5=7.2 Hz), 7.05 (m, 1H, J2′–3′=6.6 Hz, J3′–4′=6.8 Hz), 7.25 (m, 1H, J2′–4′=1.2 Hz, J3′–4′=6.8 Hz, J4′–5′=7.2 Hz), 7.57 (dd, 1H, J2′–4′=1.2 Hz, J2′–3′=6.6 Hz), 7.92 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 20.1 (1C, O=C–CH3), 25.0 (1C, –CH2–CH2–CH2–C=O), 31.6 (1C, –CH2–CH2–CH2–C=O), 37.9 (1C, –NH–CH2–CH2–CH2–), 45.6 (1C, –N–CH3), 48.3 (2C, –O=C–N–CH2–CH2–N–), 53.9 (2C, –O=C–N–CH2–CH2–N–), 115.3, 118.7, 124.3, 127.3, 129.8 and 138.3 (6C, Ar), 162.5 (1C, –NH–C=O), 167.5 (1C, CH3–C=O), 173.2 (1C, –CH2–C(=O)–O); HR-MS(ESI): (M+H+) 348.2073 (calculated 348.2068), error=1.4 ppm.
Antiepileptic Activity AssayPentylenetetrazol (PTZ), kainic acid, bicuculline, picrotoxin, 4-AP, and pilocarpine have been tested for their capacity to induce behavioral seizures in freely swimming tadpoles when bath applied. All six chemoconvulsants consistently induced similar patterns of abnormal behavior in a dose-dependent manner, characterized by convulsive clonus-like motor patterns with periods of behavioral arrest.57) Dose–response studies found 4-AP to be particularly well suitable for an experimental chemo convulsant for mice due to the rapid onset and long duration of action that reliably elicited severe seizure behavior without toxicity or lethality.
Epilepsy ModelThe 4-AP seizure test was carried out as previously described using a 7 mg/kg dose, which as confirmed in the present study produced tonic seizures in 95% of control mice. Mice were pretreated subcutaneously with either saline (control group) or compounds 30 min before receiving 4-AP. Mice that failed to exhibit tonic seizures (tonic extension of hind limbs) within 45 min after 4-AP administration were considered to be protected. The behavioral seizure that resulted from each stimulus train was graded by the following standardized scale: (behavioral seizure score: 0=no seizure, 1=seizure with mouth and facial movement, 2=seizure with head nodding, 3=seizure with forelimb clonus, 4=seizure with rearing up on hindlimbs, and 5=seizure with rearing and falling).
The Initial Anticonvulsant Evaluation of the Synthesized Compounds mice (bisexual each half) which were ignited by 4-AP were randomly assigned to 81 experimental groups (n=6). Each of compounds was provided by intraperitoneal injection with the dosage of 80 mg/kg, 160 mg/kg, 240 mg/kg, and 4-AP was provided by intraperitoneal injection after 20 min. Control group was provided with isometric the physiological saline, model group isometric the solution of 4-AP and positive control group isometric the solution of Sodium VPA.
The Test of Median Lethal Dose (LD50) mice were randomly divided into 75 groups and were injected intraperitoneal injection with different doses of biologically active compounds as specified (300 mg/kg, 360 mg/kg, 432 mg/kg, 518 mg/kg, 622 mg/kg). The number of mice that died within 3 d after administration was counted. The formula of LD50 was as follows:
Xm: logarithms of maximum doses, i: logarithms of the ratio of adjacent groups (lgr), P: mortality of the animals in each group
The Test of Median Effective Dose (ED50)Six hundreds sixteen model mice which were ignited by 4-AP (7 mg/kg) were randomly divided into 77 groups and injected intraperitoneal injection with different doses of biologically active compounds as specified (100 mg/kg, 120 mg/kg, 144 mg/kg, 173 mg/kg, 207 mg/kg). The formula of ED50 was as follows:
Xm: logarithms of maximum doses, i: logarithms of the ratio of adjacent groups (lgr), P: mortality of the animals in each group
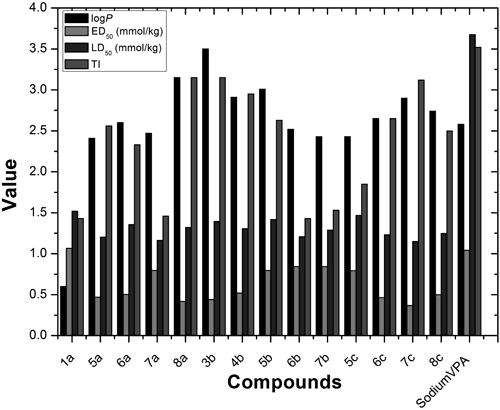
Therapeutic Index (TI)TI for each compound which was tested in ED50 and LD50, was calculated by dividing a given LD50 value evaluated in the LD50 test, by the respective ED50 value determined in the ED50 test. The TI is considered as an index of the safety and tolerability between anticonvulsant doses and toxicity.
1-Octanol/Water Partition Coefficients of the Active CompoundsLog P were determined with ChemDraw 12.0 software.
Assay of Compounds 8a, 7c and ASA Concentration in Brain TissueThe control mice were administered with ASA and a respective vehicle, and meanwhile the tested mice with 8a/7c. Mice were killed by decapitation at times chosen to coincide with those scheduled for the seizure test and brains were removed from skulls, weighed and homogenized using original Abbott buffer (2 : 1, v/w) in an Ultra-Turrax homogenizer. The concentrations of ASA, 8a and 7c were determined by HPLC using an automated Gilson (Anachem) HPLC system. The mobile phase comprised of methanol (20 mmol), acetonitrile and citric buffer (pH=3) in a ratio of 25 : 30 : 40 (v/v/v). Chromatographic separation was achieved using a Hypersil BDS-2-C18 5 µm column (Agilent Technologies). Brain homogenate samples were prepared for analysis as follows. Brain homogenate (20 µL) was pipetted into a C18 column conditioned with methanol (2×1 mL) and water (2×1 mL) and eluted with methanol (2×0.2 mL) and water (0.4 mL). Subsequently samples were centrifuged for 5 min, and the supernatant (20 µL) was transferred into the HPLC column. The limit detection of the HPLC method was 0.1 µg/mL and the within-batch and between-batch coefficients of variation were below 6%. Total compounds concentrations were expressed in µg/mL of brain supernatants as mean±S.D. of at least eight separate brain preparations.