Abstract
For indigo and its isomers epindolidione and indirubin, potential energy profiles of the excited-state intramolecular proton transfer (ESIPT) and subsequent nonradiative deactivation via conical intersection (CI) are calculated using the TDDFT and MS-CASPT2 methods. Both methods predict that indigo and indirubin exhibit a lower energy barrier for ESIPT compared to epindolidione. In addition, energy of S1/S0 CI relative to the minimum after the ESIPT has been found to be the highest for epindolidione. These findings suggest that nonradiative deactivation is more efficient in indigo and indirubin than in epindolidione, which is consistent with the experimental observation that fluorescence is quenched only in the former.
1 序論
インディゴは最も広く使われている天然染料のひとつであり,高い光安定性が特徴である.インディゴの蛍光について,量子収率は極めて低く,寿命も0.14 nsと非常に短いことが分かっている [1].この実験結果は,電子励起されたインディゴが非常に効率的な無放射失活を起こし,それが光安定性の起源となることを示唆している.
我々は以前の理論研究 [2] で,インディゴのS1 (1ππ*) 状態においてNH基からCO基への励起状態分子内プロトン移動 (ESIPT) 反応が起こることを明らかにした.さらに,プロトンが1個移動した後の構造において,S1状態とS0状態のポテンシャルエネルギー曲面間の円錐交差 (CI) を見出した.このCIが,インディゴにおける超高速無放射失活の原因であると考えられる.
一方,インディゴとその構造異性体エピンドリジオンとインディルビンは,交差共役系 (H型発色系) を持つという共通の吸光機構を示すが,その蛍光特性には大きな差異がある.エピンドリジオンの蛍光強度はインディゴより遥かに大きい [3] のに対し,インディルビンではインディゴ同様に蛍光量子収率が非常に小さい [1].
本研究の目的は,前段のような蛍光特性の違いが生じる原因を明らかにすることである.この目的のため,インディゴを含めた三種類の異性体分子について,ESIPTのポテンシャル曲線の計算およびCIの探索を実行した.
2 計算方法
S1状態の安定構造および遷移状態構造の最適化をTDDFT法 (CAM-B3LYP汎関数) で,S1・S0状態間のCI (S1/S0 CI) の構造最適化をCASSCF法で実行した.さらに,これらの最適化構造についてMS-CASPT2法でエネルギーを再計算した.CASSCFおよびMS-CASPT2計算における活性空間は (2,2) とした.
またS1状態について,1個のプロトンが移動する単プロトン移動 (SPT) 反応のポテンシャルエネルギー曲線をTDDFT/CAM-B3LYPレベルで計算した.NH間距離とOH間距離の差 (saとする) を反応座標に設定してこれを固定し,他の内部自由度をS1状態に対して最適化した.
全ての計算において,基底関数にはSapporo-DZP-2012 [4] を使用した.プログラムパッケージは,TDDFT計算にはGAMESSを,CASSCFおよびMS-CASPT2計算にはMolpro 2015.1を使用した.
3 結果と考察
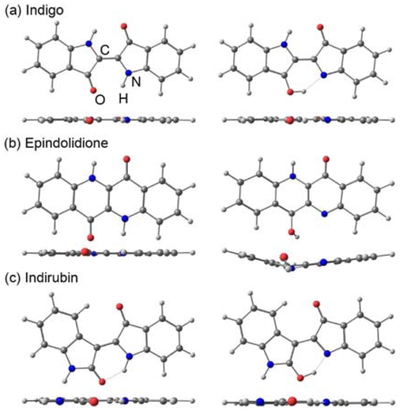
Figure 1に,ESIPT (SPT) 反応について計算したポテンシャル曲線を示す.S1 (赤実線) におけるsa < 0とsa > 0の極小点が,それぞれSPT前 (ケト体) とSPT後 (モノエノール体) の安定構造に対応する.これらの構造および遷移状態の分子構造は,いずれも平面対称性 (C2hまたはCs) を保持している.このうち,ケト体のS1安定構造をFigure 2に示す.この構造と遷移状態構造のエネルギー差から反応障壁を計算すると,TDDFT法では,インディゴで0.24 eV,エピンドリジオンで0.50 eV,インディルビンで0.16 eV と求められた.MS-CAPST2法で計算した障壁は,それぞれ0.06,0.36,0.04 eVであった.これらの結果から,インディゴとインディルビンのESIPTはエピンドリジオンよりも起こりやすいと考えられる.
また,全ての分子で,モノエノール体におけるS1/S0 CI の構造が得られた (Figure 2).インディゴとインディルビンでは平面対称性を維持するのに対し,エピンドリジオンではOH基を持つ六員環が折れ曲がった構造となった.このCIについて,ケト体のS1安定構造とのエネルギー差は,インディゴで0.66 eV,エピンドリジオンで0.70 eV,インディルビンで0.71 eVと計算された (MS-CASPT2レベル.CIのエネルギーはS1とS0の平均値).一方,モノエノール体のS1安定構造とのエネルギー差は,それぞれ0.56,1.35,0.62 eVであった.この結果は,仮にエピンドリジオンでSPTが起こっても,その後の無放射失活は他の二つよりも起こりにくいことを示唆する.
さらに,ケト体におけるエピンドリジオンの発光エネルギーは,TDDFT法で3.19 eV,MS-CASPT2法で2.71 eVと計算された.これらの計算値は蛍光波長の実験値 (484 nm, 2.56 eV [3]) とよい一致を示しており,エピンドリジオンではESIPTの起こる前に発光が生じると言える.
以上の計算結果は,三種類の分子の蛍光特性に関する実験結果と整合する.つまり,インディゴとインディルビンではESIPT,並びにCIを介した無放射失活がエピンドリジオンよりも効率的に起こり,その結果として前者二つの蛍光のみが非常に弱くなると結論づけられる.また,本研究の結果は,水素結合顔料の蛍光分子設計においてESIPTの制御が重要であることを示している.
Acknowledgment
本研究における計算の一部は,計算科学研究センター (岡崎) のスーパーコンピュータを利用して実施した.
参考文献
- [1]
J. Seixas de Melo, A. P. Moura, M. J. Melo, J. Phys. Chem. A, 108, 6975 (2004). doi:10.1021/jp049076y
- [2]
S. Yamazaki, A. L. Sobolewski, W. Domcke, Phys. Chem. Chem. Phys., 13, 1618 (2011).
, doi:10.1039/C0CP01901A21152507
- [3]
E. D. Głowacki, G. Romanazzi, C. Yumusak, H. Coskun, U. Monkowius, G. Voss, M. Burian, R. T. Lechner, N. Demitri, G. J. Redhammer, N. Sünger, G. P. Suranna, S. Sariciftci, Adv. Funct. Mater., 25, 776 (2015). doi:10.1002/adfm.201402539
- [4]
T. Noro, M. Sekiya, T. Koga, Theor. Chem. Acc., 131, 1124 (2012). doi:10.1007/s00214-012-1124-z